Vaikuttavat aineet: erytropoietiini (epoetiini alfa)
EPREX 2000 IU / ml, 4000 IU / ml, 10000 IU / ml ja 40000 IU / ml INJEKTIOLiuos, esitäytetyissä ruiskuissa
Miksi Eprexiä käytetään? Mitä varten se on?
Mitä EPREX on ja mihin sitä käytetään
EPREX sisältää vaikuttavana aineena epoetiini alfaa, proteiinia, joka stimuloi luuydintä tuottamaan enemmän punasoluja, soluja, jotka kuljettavat hemoglobiinia (happea kuljettava aine) .Epoetiini alfa on kopio ihmisen erytropoietiinista ja toimii tavalla.
- EPREXiä käytetään munuaisten vajaatoiminnan aiheuttaman oireisen anemian hoitoon.
- hemodialyysihoitoa saavilla lapsilla
- aikuisilla, jotka saavat hemodialyysiä ja peritoneaalidialyysiä
- aikuisilla, joilla on vaikea anemia ja jotka eivät ole vielä dialyysihoidossa.
Jos sinulla on munuaisten vajaatoiminta ja munuaisesi eivät tuota riittävästi erytropoietiinia (joka on välttämätöntä punasolujen tuottamiseksi), veressäsi voi olla vähän punasoluja. EPREX on määrätty stimuloimaan luuydintä tuottamaan enemmän punasoluja.
- EPREXiä käytetään anemian hoitoon, joka voi ilmetä solunsalpaajahoidon aikana kiinteiden kasvainten, pahanlaatuisen lymfooman tai multippelin myelooman (luuydinsyöpä) hoidossa, jos lääkäri katsoo, että sinun on ehkä saatava verensiirto.
- EPREXiä käytetään kohtalaisen aneemisten potilaiden hoitoon, jotka ovat ehdokkaita tallentamaan verensä ennen leikkausta, jotta se voidaan siirtää verensiirtoon leikkauksen aikana tai sen jälkeen. suurempi määrä verta.
- EPREXiä käytetään kohtalaisen aneemisten aikuispotilaiden hoitoon, joille tehdään suuri ortopedinen leikkaus (esim. Lonkan tai polven tekonivelleikkaus) verensiirtojen tarpeen vähentämiseksi.
Vasta -aiheet Milloin Eprexiä ei tule käyttää
Älä käytä EPREXiä
- Jos olet allerginen (yliherkkä) epoetiini alfalle tai EPREXin jollekin muulle aineelle (lueteltu pakkauksen sisältö ja muut tiedot)
- Jos sinulla on diagnosoitu "puhdas punasoluaplasia (luuydin ei pysty tuottamaan riittävästi punasoluja) edellisen hoidon jälkeen jollakin aineella, joka stimuloi punasolujen tuotantoa (mukaan lukien EPREX)." Katso kohta Mahdolliset haittavaikutukset.
- Jos sinulla on ongelmia hallitsemattoman korkean verenpaineen kanssa
- Edistää punasolujen tuotantoa (jotta sinulta voidaan ottaa enemmän verta), jos et voi saada oman verensiirtoja leikkauksen aikana tai sen jälkeen.
- Jos sinulle tehdään valinnainen suuri ortopedinen leikkaus (esim. Lonkan tai polven tekonivelleikkaus) ja
- on vaikea sydänsairaus
- sinulla on vakavia ongelmia suonissasi ja valtimoissasi
- sinulla on äskettäin ollut sydänkohtaus tai aivohalvaus
- ei voi ottaa verta ohentavia lääkkeitä
EPREX ei ehkä sovi sinulle. Keskustele asiasta lääkärisi kanssa. Jotkut ihmiset saattavat EPREXin käytön aikana tarvita lääkkeitä verihyytymien riskin vähentämiseksi.Jos et voi ottaa antikoagulanttilääkkeitä, älä käytä EPREXiä.
Käyttöä koskevat varotoimet Mitä sinun on tiedettävä, ennen kuin otat Eprex -valmistetta
Ole erityisen varovainen EPREXin suhteen
EPREX ja muut punasoluja stimuloivat aineet voivat lisätä verihyytymien kehittymisen riskiä kaikilla potilailla. Tämä riski voi olla suurempi, jos sinulla on muita veritulppien kehittymisen riskitekijöitä (esimerkiksi jos sinulla on aiemmin ollut veritulppa tai olet ylipainoinen, sinulla on diabetes, sinulla on sydänsairaus tai jos olet ollut immobilisoitu pitkään leikkaus tai sairaus). Kerro lääkärillesi kaikista näistä asioista. Lääkärisi auttaa sinua päättämään, onko EPREX sinulle sopiva.
On tärkeää, että keskustelet lääkärisi kanssa, jos jokin seuraavista koskee sinua.
Voit edelleen käyttää EPREXiä, mutta keskustele asiasta ensin lääkärisi kanssa.
- Jos tiedät, että sinulla on kipuja tai olet kärsinyt:
- Korkea verenpaine;
- Kouristukset tai kohtaukset
- maksasairaus;
- Anemia muista syistä;
- Porfyria (harvinainen verisairaus).
- Jos sinulla on syöpä, sinun tulee olla tietoinen siitä, että punasolujen tuotantoa stimuloivat aineet (kuten EPREX) voivat toimia kasvutekijänä ja teoriassa vaikuttaa kasvaimen etenemiseen. Verensiirto voi olla suositeltavaa yksilöllisestä tilasta riippuen. Keskustele tästä lääkärisi kanssa.
Kiinnitä erityistä huomiota aineisiin, jotka stimuloivat punasolujen tuotantoa:
EPREX kuuluu aineiden ryhmään, jotka stimuloivat punasolujen tuotantoa, kuten ihmisen erytropoieettiset tekijät. Lääkärin tulee aina kirjata käyttämänsä tuotteen tarkka nimi. Jos saat hoidon aikana samaan ryhmään kuuluvaa ainetta, mutta eri kuin EPREX, ota yhteys lääkäriisi tai apteekkiin ennen käyttöä.
Yhteisvaikutukset Mitkä lääkkeet tai elintarvikkeet voivat muuttaa Eprexin vaikutusta
EPREX ei tavallisesti vaikuta muihin lääkkeisiin, mutta kerro aina lääkärillesi, jos parhaillaan käytät tai olet äskettäin käyttänyt muita lääkkeitä, myös lääkkeitä, jotka eivät edellytä reseptiä.
Jos käytät syklosporiini -nimistä lääkettä (käytetään esimerkiksi munuaisensiirron jälkeen), lääkäri voi määrätä verikokeita syklosporiinipitoisuuksien tarkistamiseksi EPREX -hoidon aikana.
Raudan lisäravinteet ja muut anemian vastaiset tekijät voivat lisätä EPREXin tehokkuutta. Lääkäri päättää, otatko ne.
Jos olet sairaalahoidossa tai lääkärintarkastuksessa, ilmoita, että saat hoitoa EPREXillä. Tämä voi vaikuttaa muihin hoitoihin tai testituloksiin.
Varoitukset On tärkeää tietää, että:
Raskaus ja imetys
On tärkeää kertoa lääkärille, jos jokin seuraavista tiloista koskee sinua. Voit edelleen käyttää EPREXiä, mutta keskustele asiasta ensin lääkärisi kanssa.
- Jos olet raskaana tai epäilet olevasi raskaana.
- Jos imetät.
Annos, menetelmä ja antotapa Eprexin käyttö: Annostus
Käytä tätä lääkettä aina lääkärisi ohjeiden mukaan. Tarkista lääkäriltäsi, jos olet epävarma.
Lääkärisi on verikokeidesi perusteella määrittänyt, että tarvitset EPREXiä.
EPREX voidaan antaa injektiona:
- Suoneen tai putkeen laskimoon (laskimonsisäisesti)
- Ihon alle (ihon alle)
Lääkärisi päättää, miten EPREX annetaan. Pistokset antaa yleensä lääkäri, sairaanhoitaja tai terveydenhuollon ammattilainen; Jotkut ihmiset voivat oppia antamaan lääkkeen ihonalaisesti yksinään: katso ohjeet EPREXin pistämisestä itse.
- Eprexiä ei tule käyttää:
- etiketissä tai ulkopakkauksessa mainitun viimeisen käyttöpäivämäärän jälkeen
- jos tiedät tai luulet, että lääke on saattanut jäädyttää vahingossa, tai kyllä
- ja jääkaapissa oli vika
EPREX -annos perustuu kehon painoon kilogrammoina, ja lääkäri valitsee sen anemian syyn perusteella.
Lääkärisi tarkistaa verenpaineesi säännöllisesti EPREX -hoidon aikana.
Potilaat, joilla on munuaisten vajaatoiminta
- Hemoglobiiniarvot pidetään 10–12 g / dl, koska korkeampi hemoglobiinipitoisuus voi lisätä tromboosin ja kuoleman riskiä.
- Tavallinen EPREX -aloitusannos aikuisille tai lapsille on 50 kansainvälistä yksikköä (IU) painokiloa (/ kg) kohden 3 kertaa viikossa.
- Peritoneaalidialyysihoitoa saaville potilaille lääke voidaan antaa kahdesti viikossa.
- EPREX annetaan laskimonsisäisesti (laskimoon tai putkeen laskimoon) aikuisille ja lapsille. Jos laskimonsisäinen reitti (laskimo tai putki laskimoon) ei ole käytettävissä, lääkäri voi päättää, pistetäänkö EPREX ihon alle (ihon alle). Sisältää dialyysipotilaat ja potilaat, jotka eivät ole vielä dialyysihoidossa.
- Lääkärisi käy säännöllisesti verikokeissa tarkistaaksesi, reagoiko anemiasi, ja voi muuttaa annosta, yleensä enintään kerran 4 viikossa.
- Kun anemia on korjattu, lääkäri jatkaa verikokeidesi säännöllistä tarkistamista.
- Jos saat EPREX -hoitoa pidemmillä annosväleillä (useammin kuin kerran viikossa), et ehkä pysty ylläpitämään hemoglobiinitasojasi riittävästi ja saatat joutua lisäämään EPREX -annostasi tai sen antotiheyttä.
- Hoidon tehostamiseksi raudan lisäravinteet voivat olla hyödyllisiä sinulle ennen EPREX -hoitoa ja sen aikana.
- Jos saat hemodialyysihoitoa, kun aloitat EPREX -hoidon, dialyysihoitoa on ehkä muutettava. Lääkäri päättää.
Aikuiset potilaat, jotka saavat kemoterapiaa
- Lääkärisi voi aloittaa EPREX -hoidon, jos hemoglobiinitasosi on 10 g / dl tai vähemmän.
- Hemoglobiiniarvot pidetään 10–12 g / dl, koska korkeampi hemoglobiinipitoisuus voi lisätä tromboosin ja kuoleman riskiä.
- Aloitusannos on 150 IU painokiloa kohti 3 kertaa viikossa tai 450 IU painokiloa kohti kerran viikossa.
- EPREX annetaan ihonalaisena injektiona.
- Lääkärisi käy säännöllisesti verikokeissa ja voi muuttaa annosta sen mukaan, miten EPREX -hoitoon saat vasteen.
- Hoidon tehostamiseksi raudan lisäravinteesta voi olla sinulle hyötyä ennen EPREX -hoitoa ja sen aikana.
- EPREX -hoitoa jatketaan yleensä yhden kuukauden ajan solunsalpaajahoidon päättymisen jälkeen.
Aikuiset potilaat, jotka tallentavat oman verensä
- Tavanomainen annos on 600 IU painokiloa kohti 2 kertaa viikossa.
- EPREX annetaan laskimoon heti veren asettamisen jälkeen 3 viikon ajan ennen leikkausta.
- Hoidon tehostamiseksi raudan lisäravinteesta voi olla sinulle hyötyä ennen EPREX -hoitoa ja sen aikana.
Aikuispotilaat, jotka ovat ehdokkaita suuriin ortopedisiin leikkauksiin
- Suositeltu annos on 600 IU painokiloa kohti kerran viikossa.
- EPREX annetaan ihonalaisena injektiona joka viikko kolmen viikon ajan ennen leikkausta ja leikkauspäivänä.
- Jos leikkausta edeltävää aikaa on tarpeen lyhentää, sinulle annetaan päivittäinen annos 300 IU / kg leikkausta edeltävien 10 päivän aikana, leikkauspäivänä ja 4 päivää leikkauksen jälkeen.
- Jos verikokeet ennen leikkausta paljastavat liian korkeat hemoglobiiniarvot, hoito lopetetaan.
- Hoidon tehostamiseksi raudan lisäravinteet voivat olla hyödyllisiä sinulle ennen EPREX -hoitoa ja sen aikana.
Ohjeet EPREXin pistämiseen itse
- Hoidon alussa lääkäri tai sairaanhoitaja antaa tavallisesti EPREXiä, minkä jälkeen lääkäri voi ehdottaa, että sinä (tai hoitajasi) opettelet pistoksen (ihonalaisesti).
- Älä yritä pistää itseäsi, jos lääkäri tai sairaanhoitaja ei ole kertonut sinulle kuinka.
- Noudata aina lääkärin tai sairaanhoitajan antamia ohjeita.
- Käytä EPREXiä vain, jos se on säilytetty oikein - katso kohta EPREXin säilyttäminen
- Ota Eprex-ruisku jääkaapista ennen käyttöä ja anna sen lämmetä huoneenlämpöiseksi. Se kestää yleensä 15-30 minuuttia.
Vedä yksi kerta-annos EPREXiä jokaisesta esitäytetystä ruiskusta.
EPREX on annettava yksin eikä sitä saa sekoittaa muiden injektionesteiden kanssa.
Älä ravista esitäytettyjä EPREX-ruiskuja. Pitkäaikainen voimakas ravistelu voi vahingoittaa tuotetta. Älä käytä tuotetta, jos sitä on ravistettu voimakkaasti.
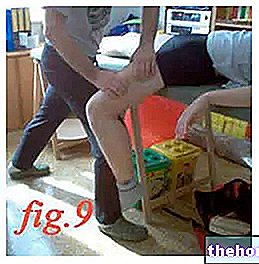
Kuinka pistät itseäsi esitäytetyillä ruiskuilla
Esitäytetyt ruiskut on varustettu neulan turvalaitteella PROTECS ™, joka estää neulan tarttumisen käytön jälkeen. Tämä on ilmoitettu pakkauksessa.
- Poista esitäytetty ruisku jääkaapista ennen käyttöä. Nesteen on lämmettävä huoneenlämpötilaan. Älä poista neulansuojusta odottaessasi huoneenlämpöistä.
- Tarkista esitäytetty ruisku, että se on oikea annos, että se ei ole vanhentunut, ettei se ole vaurioitunut ja että neste on kirkasta eikä jäätynyt.
- Valitse pistoskohta. Sopivimmat pistoskohdat ovat reisi ja vatsa, lukuun ottamatta napaa ympäröivää aluetta. Vaihda pistoskohta joka kerta.
- Käsien pesuun. Desinfioi pistoskohta antiseptisellä pyyhkeellä.
- Pidä esitäytettyä ruiskua ruiskuputkesta kiinni peitetty neula ylöspäin.
- Älä pidä kiinni männän päästä, männästä, neulan suojuksesta tai neulan suojuksesta.
- Älä vedä mäntää taaksepäin missään olosuhteissa
- Älä irrota esitäytetyn ruiskun neulansuojusta ennen kuin olet valmis antamaan EPREX-valmistetta
- Poista ruiskun neulan suojakorkki pitämällä sitä rungosta ja vetämällä korkista kiertämättä sitä.Älä työnnä mäntää, kosketa neulaa tai ravista ruiskua. - Älä kosketa turvalaitteen aktivointiklipsejä estääksesi neulan ennenaikaisen peittämisen neulansuojuksella
- Nosta ihoa peukalon ja etusormen väliin puristamatta sitä liikaa.
- Työnnä neula kokonaan sisään. Lääkärisi tai sairaanhoitaja näyttää sinulle kuinka.
- Työnnä mäntää peukalolla, kunnes oikea määrä annosta vastaavaa nestettä on ruiskutettu. Työnnä hitaasti ja tasaisesti pitäen ihoa koholla. PROTECS ™ -neulan turvalaite ei aktivoidu ennen kuin pistos on annettu. Kuulet "napsahduksen", kun PROTECS ™ -neulan turvalaite on aktivoitu.
- Kun mäntä on saavuttanut iskunsa lopun, vedä neula ulos ja vapauta iho.
- Poista peukalo hitaasti männästä, jotta neula peittyy kokonaan turvalaitteella.
- Pistoksen lopussa pistoskohdassa voi olla pieni määrä verta. Tämä on normaalia. Voit desinfioida pistoskohdan painamalla antiseptistä tyynyä muutaman sekunnin ajan.
- Hävitä käytetyt ruiskut turvalliseen astiaan - katso kohta Eprexin säilyttäminen
Yliannostus Mitä tehdä, jos olet ottanut liian paljon Eprexiä?
Jos käytät enemmän EPREXiä kuin sinun pitäisi
Kerro heti lääkärille tai sairaanhoitajalle, jos epäilet käyttäneesi liikaa EPREXiä. Yliannostuksen sivuvaikutukset ovat epätodennäköisiä.
Jos unohdat pistää EPREXin
Suorita seuraava pistos heti kun muistat.Jos seuraava pistos on vuorokauden sisällä, jätä unohtunut annos väliin ja jatka tavanomaista aikataulua. Älä kaksinkertaista injektioita.
Jos olet hepatiitti C -potilas ja saat interferonia ja ribaviriinia
Sinun on otettava yhteys lääkäriisi, koska epoetiini alfa -yhdistelmän ja interferonin ja ribaviriinin yhdistelmä on harvinaisissa tapauksissa johtanut tehon menetykseen ja vakavan anemian muodon, nimeltään puhdas punasoluaplasia (PRCA), kehittymiseen. EPREXiä ei ole hyväksytty hepatiitti C: hen liittyvän anemian hoitoon.
Jos sinulla on kysymyksiä tämän tuotteen käytöstä, käänny lääkärin, sairaanhoitajan tai apteekkihenkilökunnan puoleen.
Sivuvaikutukset Mitkä ovat Eprexin sivuvaikutukset
Kuten kaikki lääkkeet, myös EPREX voi aiheuttaa haittavaikutuksia. Kaikki eivät kuitenkaan niitä saa. Kerro heti lääkärille tai sairaanhoitajalle, jos jokin alla luetelluista vaikutuksista ilmenee.
Hyvin yleiset haittavaikutukset
Niitä esiintyy useammalla kuin yhdellä kymmenestä potilaasta, jotka käyttävät EPREXiä
- Ripuli
- Pahoinvointi vatsassa
- Hän vetäytyi
- Kuume
- Hengitysteiden tukkoisuutta, kuten nenän tukkoisuutta ja kurkkukipua, on raportoitu potilailla, joilla on munuaissairaus ja jotka eivät ole vielä dialyysihoidossa.
Yleiset haittavaikutukset
Näitä esiintyy enintään yhdellä potilaalla 10: stä, jotka käyttävät EPREXiä
- Verenpainearvojen nostaminen. Päänsärky (varsinkin jos äkillinen, akuutti ja migreenin kaltainen) tai sekavuus tai kouristukset. Nämä voivat olla merkkejä äkillisestä verenpaineen noususta, joka vaatii välitöntä hoitoa. Verenpaineen nousu voi vaatia hoitoa. Lääkkeillä (tai korkean verenpaineen hoitoon jo käyttämiesi lääkkeiden annoksen muuttaminen).
- Veritulpat (mukaan lukien syvä laskimotukos ja embolia), jotka voivat vaatia kiireellistä hoitoa. Oireita voivat olla rintakipu, hengityksen vinkuminen, alaraajojen kivulias turvotus ja punoitus, yleensä jaloissa.
- yskä.
- Ihon ärsytys, joka voi johtua allergisesta reaktiosta.
- Luu- tai lihaskipu.
- Flunssan kaltainen oireyhtymä, kuten päänsärky, nivelkivut, heikkouden tunne, vilunväristykset, väsymys ja huimaus. Nämä reaktiot ovat yleisempiä hoidon alussa.Jos sinulla ilmenee näitä oireita pistoksena laskimoon, hitaampi antaminen voi auttaa välttämään niitä tulevaisuudessa.
- Punoitus, polttaminen ja kipu pistoskohdassa.
- Turvotus nilkoissa, jaloissa tai sormissa.
Melko harvinaiset haittavaikutukset
Näitä esiintyy enintään yhdellä 100: sta EPREXiä käyttävästä potilaasta
- Korkea veren kaliumpitoisuus, joka voi aiheuttaa epänormaalin sydämen rytmin (tämä on hyvin yleinen haittavaikutus dialyysipotilailla).
- Kouristukset
- Nenän tai hengitysteiden tukkoisuus
Hyvin harvinaiset haittavaikutukset
Näitä esiintyy enintään yhdellä potilaalla 10000: sta, jotka käyttävät EPREXiä
- Puhtaan punasoluaplasian (PRCA) oireet. PRCA tarkoittaa kyvyttömyyttä tuottaa tarpeeksi punasoluja luuytimessä. PRCA voi aiheuttaa vakavan ja äkillisen anemian, jonka oireita ovat:
- epätavallinen väsymys,
- huimauksen tunne,
- hengenahdistus.
PRCA: ta on todettu hyvin harvoin erityisesti munuaissairailla potilailla kuukausien tai vuosien EPREX -hoidon ja muiden punasolujen tuotantoa stimuloivien aineiden jälkeen. Pienten verisolujen (verihiutaleiden), tavallisesti verihyytymien muodostumiseen osallistuvien verihiutaleiden, määrä voi nousta, erityisesti hoidon alussa. Lääkäri tarkistaa tämän.
Jos saat hemodialyysihoitoa:
- Dialyysisunttiin voi muodostua verihyytymiä (tromboosi).Tämä voi tapahtua helpommin, jos sinulla on alhainen verenpaine (hypotensio) tai jos sinulla on ongelmia fistelisi kanssa.
- Hemodialyysijärjestelmään voi myös muodostua verihyytymiä.Lääkäri voi päättää suurentaa hepariiniannosta dialyysin aikana.
Kerro heti lääkärille tai sairaanhoitajalle, jos huomaat jonkin näistä vaikutuksista tai jos havaitset muita vaikutuksia EPREX -hoidon aikana.
Jos havaitset sellaisia haittavaikutuksia, joita ei ole mainittu tässä pakkausselosteessa, kerro niistä lääkärille, sairaanhoitajalle tai apteekkihenkilökunnalle.
Niille, jotka harjoittavat urheilutoimintaa: lääkkeen käyttö ilman terapeuttista tarvetta on dopingia ja voi joka tapauksessa määrittää positiivisen dopingtestin.
Vanhentuminen ja säilyttäminen
Ei lasten ulottuville eikä näkyville.
Älä käytä tätä lääkettä pakkauksessa ja etiketissä mainitun viimeisen käyttöpäivämäärän EXP jälkeen. Viimeinen käyttöpäivämäärä on mainitun kuukauden viimeinen päivä.
EPREX on säilytettävä jääkaapissa (2 ° C - 8 ° C).
EPREX voidaan ottaa jääkaapista ja säilyttää huoneenlämmössä (enintään 25 ° C) enintään 3 päivää. Kun esitäytetty ruisku on otettu pois jääkaapista ja se on saavuttanut huoneenlämpötilan (enintään 25 ° C), se on käytettävä 3 päivän kuluessa tai hävitettävä.
Sitä ei saa jäätyä tai ravistaa
Säilytä alkuperäisessä pakkauksessa valolta suojaamiseksi.
Älä käytä tätä lääkettä, jos sinetti on rikki tai jos liuos on värjäytynyttä tai siinä on hiukkasia. Jos näitä ehtoja havaitaan, hävitä lääke
Lääkkeitä ei tule heittää viemäriin eikä hävittää talousjätteiden mukana. Kysy apteekista, kuinka heittää pois käyttämättömät lääkkeet. Tämä auttaa suojelemaan ympäristöä.
Pakkauksen sisältö ja muuta tietoa
Mitä Eprex sisältää
Vaikuttava aine on: epoetiini alfa (määrä alla olevasta taulukosta).
Muut aineet ovat: Polysorbaatti 80, natriumkloridi, yksiemäksinen natriumfosfaattidihydraatti, kaksiemäksinen natriumfosfaattidihydraatti, glysiini ja injektionesteisiin käytettävä vesi. Tämä lääke sisältää alle 1 mmol natriumia (23 mg) per annos, joten se on käytännössä natriumiton.
Lääkevalmisteen kuvaus ja pakkaus
EPREX on injektioneste, liuos esitäytetyssä ruiskussa. Esitäytetyt ruiskut on varustettu PROTECS ™ -neulansuojalaitteella (katso alla oleva taulukko) EPREX on kirkas ja väritön liuos.
Kaikkia pakkauskokoja ei välttämättä ole myynnissä
Lähdepakkaus: AIFA (Italian lääkevirasto). Sisältö julkaistu tammikuussa 2016. Esitetyt tiedot eivät välttämättä ole ajan tasalla.
Jotta saat käyttöösi uusimman version, on suositeltavaa käyttää AIFA: n (Italian Medicines Agency) verkkosivustoa. Vastuuvapauslauseke ja hyödyllistä tietoa.
01.0 LÄÄKEVALMISTEEN NIMI
EPREX 10000 IU / ml injektioneste, liuos esitäytetyssä ruiskussa
02.0 LAADULLINEN JA MÄÄRÄLLINEN KOOSTUMUS
Epoetiini alfa 10000 IU / ml (84,0 μg / ml), joka on valmistettu yhdistelmä -DNA -tekniikalla kiinalaisen hamsterin munasarjasoluissa (CHO).
Yksi 0,3 ml: n esitäytetty ruisku sisältää 3000 IU (25,2 mcg) epoetiini alfaa
Yksi 0,4 ml: n esitäytetty ruisku sisältää 4000 IU (33,6 mcg) epoetiini alfaa
Yksi 0,5 ml: n esitäytetty ruisku sisältää 5000 IU (42,0 mcg) epoetiini alfaa
Yksi 0,6 ml: n esitäytetty ruisku sisältää 6000 IU (50,4 mikrogrammaa) epoetiini alfaa
Yksi 0,8 ml: n esitäytetty ruisku sisältää 8000 IU (67,2 mcg) epoetiini alfaa
Yksi 1,0 ml: n esitäytetty ruisku sisältää 10000 IU (84,0 mcg) epoetiini alfaa
Tämä lääkevalmiste sisältää alle 1 mmol (23 mg) natriumia annosta kohti, eli se on olennaisesti "natriumiton".
Täydellinen apuaineluettelo, katso kohta 6.1
03.0 LÄÄKEMUOTO
Injektioneste, liuos, esitäytetty ruisku
Kirkas, väritön liuos
04.0 KLIINISET TIEDOT
04.1 Käyttöaiheet
EPREX on tarkoitettu krooniseen munuaisten vajaatoimintaan (CRI) liittyvän oireisen anemian hoitoon:
• 1–18 -vuotiaille aikuisille ja lapsipotilaille, jotka saavat hemodialyysiä, ja aikuispotilaille, jotka ovat peritoneaalidialyysissä.
• aikuispotilaille, joilla on munuaisten vajaatoiminta ja jotka eivät ole vielä dialyysihoidossa vaikean munuaisperäisen anemian hoitoon, johon liittyy kliinisiä oireita potilailla.
EPREX on tarkoitettu aikuispotilaille, jotka saavat solunsalpaajahoitoa kiinteiden kasvainten, pahanlaatuisen lymfooman tai multippelin myelooman hoitoon ja joilla on verensiirtoriski potilaan yleistilan (sydän- ja verisuonitilanne, kemoterapian alussa esiintynyt anemia) perusteella anemian ja verensiirron tarpeesta.
EPREX on tarkoitettu aikuispotilaille, jotka ovat mukana autologisen veren määrän lisäämistä varten. mmol / l], ei raudanpuutetta), jos veren varastointimenettelyt eivät ole käytettävissä tai ne eivät ole riittäviä, jos kyseessä on suuri valinnainen leikkaus, joka vaatii suuren määrän verta (4 tai enemmän yksikköä naista kohden tai 5 tai enemmän yksikköä miehillä).
EPREX on tarkoitettu aikuispotilaille, joilla ei ole raudanpuutetta ennen suurta elektiivistä ortopedistä leikkausta, jolla uskotaan olevan suuri verensiirtokomplikaatioiden riski, allogeenisille verensiirroille altistumisen vähentämiseksi. Käyttö on rajoitettava potilaisiin, joilla on kohtalainen anemia (hemoglobiinipitoisuus) välillä Hb10-13 g / dl), joille ei ole saatavilla autologista verenluovutusohjelmaa ja joille odotetaan kohtalaista verenhukkaa (900-1800 ml).
04.2 Annostus ja antotapa
Annostus
Kaikki muut anemian syyt (rauta-, folaatti- tai B12 -vitamiinin puutos, alumiinimyrkytys, infektiot tai tulehdukset, verenhukka, hemolyysi ja minkä tahansa alkuperän luuydinfibroosi) ennen epoetiini alfa -hoidon aloittamista ja annoksen suurentamista päätettäessä. Optimaalisen vasteen varmistamiseksi epoetiini alfaan on varmistettava riittävät rautavarastot ja tarvittaessa annettava rautaa (ks. Kohta 4.4).
Oireisen anemian hoito aikuispotilailla, joilla on krooninen munuaisten vajaatoiminta (CRI)
Anemian oireet ja seuraukset voivat vaihdella sukupuolen, iän ja jatkuvien samanaikaisten sairauksien mukaan; lääkärin on arvioitava yksittäisen potilaan kliininen tila.
Haluttu hemoglobiinipitoisuus on 10 g / dl - 12 g / dl (6,2 - 7,5 mmol / l). EPREX on annettava siten, että hemoglobiinipitoisuus on enintään 12 g / dl (7,5 mmol / l). Hemoglobiinin nousua yli 2 g / dl (1,25 mmol / l) neljän viikon aikana tulee välttää. Jos näin tapahtuu, annosta on muutettava asianmukaisesti.
Potilaan sisäisen vaihtelun vuoksi potilaalla voidaan toisinaan havaita halutun hemoglobiinipitoisuuden ylä- ja alapuolella olevia hemoglobiiniarvoja. Tätä vaihtelua on hallittava muuttamalla annosta ottaen huomioon hemoglobiinipitoisuusalue 10 g / dl (6,2 mmol / l) - 12 g / dl (7,5 mmol / l).
Hemoglobiinitasoa, joka on jatkuvasti yli 12 g / dl (7,5 mmol), tulee välttää. Jos hemoglobiini nousee yli 2 g / dl (1,25 mmol / l) kuukaudessa tai jos hemoglobiinitaso ylittää jatkuvasti 12 g / dl (7,5 mmol), vähennä EPREX -annosta 25%. Jos hemoglobiini ylittää 13 g / dl ( 8,1 mmol / l), keskeytä hoito, kunnes se palautuu arvoon 12 g / dl (7,5 mmol / l), ja jatka sitten EPREX -hoitoa 25% pienemmillä annoksilla kuin edelliset.
Potilaita on seurattava tarkasti sen varmistamiseksi, että käytetään pienintä hyväksyttyä tehokasta EPREX -annosta anemian ja siihen liittyvien oireiden riittävän hallinnan varmistamiseksi pitämällä hemoglobiinipitoisuus alle tai yhtä suurena kuin 12 g / dl (7,5 mmol / l).
Varovaisuutta on noudatettava ESA -annoksen suurentamisessa potilailla, joilla on krooninen munuaisten vajaatoiminta.Potilaille, joilla on heikko hemoglobiinivaste ESA: lle, on etsittävä vaihtoehtoinen syy heikkoon vasteeseen (ks. Kohdat 4.4 ja 5.1).
EPREX -hoito on jaettu kahteen vaiheeseen - korjausvaihe ja ylläpitovaihe.
Aikuiset hemodialyysipotilaat
Hemodialyysipotilailla, joille laskimoyhteys on helposti saatavilla, suonensisäinen antoreitti on parempi.
Korjausvaihe :
Aloitusannos on 50 IU / painokilo 3 kertaa viikossa.
Tarvittaessa suurenna tai pienennä annosta 25 IU / kg (3 kertaa viikossa), kunnes haluttu hemoglobiinipitoisuus on välillä 10 g / dl - 12 g / dl (6,2 - 7,5 mmol / l) (tämä on tehtävä vähitellen vähintään neljän viikon välein).
- Huoltovaihe :
Suositeltu kokonaisviikkoannos on 75 IU / kg - 300 IU / kg.
Annosta on muutettava asianmukaisesti, jotta hemoglobiiniarvot pidetään halutun hemoglobiinipitoisuuden sisällä 10 g / dl - 12 g / dl (6,2 - 7,5 mmol / l).
Potilaat, joilla on hyvin alhainen hemoglobiinitaso (8 g / dl tai> 5 mmol / l).
Aikuiset munuaisten vajaatoimintaa sairastavat potilaat, jotka eivät ole vielä dialyysihoidossa
Potilaille, joilla laskimoyhteyttä ei ole helposti saatavilla, EPREX voidaan antaa ihon alle.
Korjausvaihe
Aloitusannos on 50 IU / painokilo 3 kertaa viikossa, jota seuraa tarvittaessa annoksen nostaminen 25 IU / kg (3 kertaa viikossa), kunnes haluttu hemoglobiinipitoisuus on saavutettu (tämä on tehtävä asteittain. vähintään neljän viikon välein).
Huoltovaihe
Ylläpitovaiheen aikana EPREX voidaan antaa joko 3 kertaa viikossa ja, jos sitä annetaan ihon alle, kerran viikossa tai kerran kahdessa viikossa.
Annos ja annostusvälit on säädettävä oikein, jotta hemoglobiiniarvot pysyvät halutulla tasolla: Hb 10-12 g / dl (6,2-7,5 mmol / l). Annosvälin pidentäminen saattaa vaatia annoksen suurentamista.
Suurin annos ei saa ylittää 150 IU / kg 3 kertaa viikossa, 240 IU / kg (korkeintaan 20000 IU) kerran viikossa tai 480 IU / kg (enintään 40000 IU) joka toinen viikko.
Peritoneaalidialyysissä olevat aikuispotilaat
Potilaille, joille laskimoyhteyttä ei ole helposti saatavilla, EPREX voidaan antaa ihon alle.
Korjausvaihe
Aloitusannos on 50 IU / kg kahdesti viikossa.
Huoltovaihe
Suositeltu ylläpitoannos on 25 IU / kg - 50 IU / kg kahdesti viikossa jaettuna kahteen yhtä suureen annokseen.
Annosta on muutettava asianmukaisesti, jotta hemoglobiiniarvot pysyvät halutulla tasolla: hemoglobiini on välillä 10 g / dl ja 12 g / dl (6,2-7,5 mmol / l).
Aikuispotilaiden, joilla on kemoterapian aiheuttama anemia, hoito
Anemian oireet ja seuraukset voivat vaihdella iän, sukupuolen ja taudin yleisen tilanteen mukaan; lääkärin on arvioitava yksittäisen potilaan kliininen tila.
EPREXiä tulee antaa aneemisille potilaille, esim. hemoglobiinipitoisuus ≤10 g / dl (6,2 mmol / l).
Aloitusannos on 150 IU / kg ihon alle 3 kertaa viikossa.
Vaihtoehtoisesti EPREX voidaan antaa ihon alle aloitusannoksella 450 IU / kg kerran viikossa.
Annosta on muutettava asianmukaisesti, jotta hemoglobiiniarvot pysyvät halutulla tasolla: hemoglobiini on välillä 10 g / dl ja 12 g / dl (6,2-7,5 mmol / l).
Potilaan sisäisen vaihtelun vuoksi potilaalla voidaan toisinaan havaita halutun hemoglobiinipitoisuuden ylä- ja alapuolella olevia hemoglobiiniarvoja. Tätä vaihtelua on hallittava muuttamalla annosta ottaen huomioon haluttu hemoglobiinipitoisuusalue 10 g / dl (6,2 mmol / l) - 12 g / dl (7,5 mmol / l).
Hemoglobiinipitoisuutta, joka on jatkuvasti yli 12 g / dl (7,5 mmol), tulee välttää. Alla on ohjeet asianmukaisen annoksen säätämisestä, jos hemoglobiini saavuttaa pitoisuudet yli 12 g / dl (7,5 mmol).
Jos 4 viikon hoidon jälkeen hemoglobiinipitoisuus on noussut vähintään 1 g / dl (0,62 mmol / l) tai retikulosyyttimäärä on noussut ≥ 40 000 solulla / μl lähtötasosta, annoksen tulee pysyä 150 IU / kg 3 kertaa viikossa tai 450 IU / kg kerran viikossa.
Jos hemoglobiinipitoisuuden nousu on
Jos hemoglobiinipitoisuuden nousu oli
Annoksen muuttaminen hemoglobiinipitoisuuden pitämiseksi välillä 10 g / dl - 12 g / dl
Jos hemoglobiinipitoisuus nousee yli 2 g / dl (1,25 mmol / l) kuukaudessa tai jos hemoglobiini ylittää 12 g / dl (7,5 mmol / l), pienennä EPREX -annosta noin 25-50%.
Jos hemoglobiinipitoisuus ylittää 13 g / dl (8,1 mmol / l), keskeytä hoito, kunnes pitoisuus laskee alle 12 g / dl (7,5 mmol / l), ja jatka sitten EPREX -hoitoa 25% pienemmällä annoksella kuin edellinen annos.
Suositeltu annos on kuvattu seuraavassa kaaviossa:
Potilaita on seurattava tarkasti sen varmistamiseksi, että pienintä hyväksyttyä annosta erytropoieesia stimuloivia aineita (ESA) käytetään anemian oireiden riittävään hallintaan.
EPREX -hoitoa on jatkettava kuukauden ajan kemoterapian päättymisen jälkeen.
Aikuisten potilaiden hoito, jotka ovat ehdokkaita leikkaukseen, joka on osa autologista verenluovutusohjelmaa
Lievästi aneemisia potilaita (hematokriitti välillä 33--39%), jotka tarvitsevat 4 tai useampia veriyksiköitä, on annettava 600 IU / kg EPREX-valmistetta laskimoon kahdesti viikossa kolmen viikon ajan ennen leikkausta.
EPREX tulee antaa verenluovutusmenettelyn päätyttyä.
Suurten elektiivisten ortopedisten leikkausten ehdokkaiden hoito aikuispotilailla
Suositeltu annos on 600 IU / kg EPREXiä ihon alle, kerran viikossa kolmen viikon ajan ennen leikkausta (-21 päivää, -14 päivää ja -7 päivää) ja leikkauspäivänä.
Jos lääketieteellinen tarve lyhentää leikkausta edeltävää odotusaikaa alle kolmeen viikkoon, annos 300 IU / kg EPREXiä annetaan ihon alle päivittäin 10 peräkkäisen päivän ajan. Ennen leikkausta, leikkauspäivänä ja 4 päivää välittömästi sen jälkeen.
Jos hemoglobiini saavuttaa 15 g / dl tai enemmän ennen leikkausta, EPREX -hoito on lopetettava eikä muita annoksia saa antaa.
Pediatriset potilaat
Oireisen anemian hoito hemodialyysipotilailla, joilla on krooninen munuaisten vajaatoiminta
Anemian oireet ja seuraukset voivat vaihdella iän, sukupuolen ja jatkuvien samanaikaisten sairauksien mukaan; lääkärin on arvioitava yksittäisen potilaan kliininen tila.
Lapsipotilailla haluttu hemoglobiinipitoisuus on 9,5 g / dl - 11 g / dl (5,9 - 6,8 mmol / l). EPREX on annettava siten, että hemoglobiinipitoisuus ei ylitä 11 g / dl (6,8 mmol / l). Hemoglobiinin nousua yli 2 g / dl (1,25 mmol / l) neljän viikon aikana tulee välttää. Jos näin tapahtuu, annosta on muutettava asianmukaisesti.
Potilaita tulee seurata tarkasti sen varmistamiseksi, että käytetään pienintä sallittua EPREX -annosta anemian ja siihen liittyvien oireiden riittävän hallinnan varmistamiseksi.
EPREX -hoito on jaettu kahteen vaiheeseen: korjausvaihe ja ylläpitovaihe.
Hemodialyysihoitoa saavilla pediatrisilla potilailla, joille laskimonsisäinen yhteys on jo saatavilla, suonensisäinen antaminen on suositeltavaa.
Korjausvaihe:
Aloitusannos on 50 IU / kg suonensisäisesti 3 kertaa viikossa.
Tarvittaessa suurenna tai pienennä annosta 25 IU / kg (3 kertaa viikossa), kunnes haluttu hemoglobiinipitoisuus on välillä 9,5 g / dl - 11 g / dl (5,9 - 6, 8 mmol / l) (tämän pitäisi tapahtua) vähitellen vähintään neljän viikon välein).
Huoltovaihe :
Annosta on muutettava asianmukaisesti, jotta hemoglobiiniarvot pidetään halutun hemoglobiinipitoisuuden sisällä 9,5 g / dl - 11 g / dl (5,9 - 6,8 mmol / l).
Yleensä alle 30 kg painavat lapset tarvitsevat suurempia ylläpitoannoksia kuin yli 30 kg painavat lapset ja aikuiset.
Pediatriset potilaat, joiden hemoglobiinipitoisuus on hyvin alhainen (6,8 g / dl tai> 4,25 mmol / l).
Kemoterapian aiheuttamaa anemiaa sairastavien lapsipotilaiden hoito.
EPREXin turvallisuutta ja tehoa solunsalpaajahoitoa saavilla lapsipotilailla ei ole varmistettu.
Lasten kirurgisten potilaiden hoito, jotka osallistuvat autologiseen luovutusohjelmaan
EPREXin turvallisuutta ja tehoa lapsipotilailla ei ole varmistettu.
Tietoja ei ole saatavilla.
Suurta elektiivistä ortopedista leikkausta odottavien lapsipotilaiden hoito
EPREXin turvallisuutta ja tehoa lapsipotilailla ei ole varmistettu.
Tietoja ei ole saatavilla.
Antotapa
Ole varovainen ennen lääkkeen käsittelyä tai antamista.
Anna EPREX -ruiskun levätä ennen käyttöä, kunnes se saavuttaa huoneenlämmön.Tämä kestää yleensä 15--30 minuuttia.
Oireisen anemian hoito aikuispotilailla, joilla on krooninen munuaisten vajaatoiminta
Kroonista munuaisten vajaatoimintaa sairastavilla potilailla, joille laskimonsisäinen pääsy on normaalisti saatavilla (hemodialyysipotilaat), on suositeltavaa antaa EPREX -valmistetta laskimoon.
Jos laskimonsisäistä pääsyä ei ole helposti saatavilla (potilaat, jotka eivät ole vielä dialyysihoidossa ja potilaat, jotka saavat peritoneaalidialyysiä), EPREX voidaan antaa ihon alle.
Aikuispotilaiden, joilla on kemoterapian aiheuttama anemia, hoito.
EPREX tulee antaa ihon alle.
Aikuisten kirurgisten potilaiden hoito, jotka osallistuvat autologiseen luovutusohjelmaan
EPREX on annettava suonensisäisesti.
Suurten elektiivisten ortopedisten leikkausten hoitoon tarkoitettujen aikuispotilaiden hoito
EPREX tulee antaa ihon alle.
Oireisen anemian hoito lapsipotilailla, joilla on krooninen munuaisten vajaatoiminta hemodialyysihoidossa
Kroonista munuaisten vajaatoimintaa sairastavilla pediatrisilla potilailla, joille laskimonsisäinen yhteys on jo saatavilla (hemodialyysipotilaat), EPREX -valmisteen laskimonsisäinen anto on suositeltavaa.
Laskimoon
Annos kestää vähintään 1-5 minuuttia kokonaisannoksesta riippuen.
Hemodialyysipotilaille bolusinjektio voidaan antaa dialyysin aikana riittävän laskimoyhteyden kautta dialyysilinjalla.Vaihtoehtoisesti injektio voidaan antaa dialyysin lopussa, fistelin kautta, minkä jälkeen annetaan 10 ml fysiologista liuosta, joka huuhtelee kulkureitit ja varmistaa tuotteen tyydyttävän pääsyn verenkiertoon.
Potilailla, joilla on esiintynyt flunssan kaltaisia reaktioita, suositellaan hitaampaa antamista (ks. Kohta 4.8).
Älä anna EPREXiä infuusiona laskimoon tai liuoksena muiden lääkkeiden kanssa.
Ihonalainen anto
Yleensä 1 ml: n enimmäismäärää kussakin pistoskohdassa ei saa ylittää. Suuremmille määrille on valittava useampi kuin yksi pistoskohta.
Injektiot tulee tehdä raajoihin tai vatsan etuseinään.
Jos lääkäri uskoo, että potilas tai hoitaja pystyy antamaan EPREX -valmisteen ihon alle turvallisesti ja asianmukaisesti, on annettava ohjeet oikeasta annoksesta ja antamisesta.
Kuten muidenkin injektoitavien valmisteiden kohdalla, tarkista, ettei liuoksessa ole hiukkasia tai värimuutoksia.
04.3 Vasta -aiheet
Yliherkkyys vaikuttavalle aineelle tai apuaineille.
Potilaita, joille kehittyy puhdas punasoluaplasia (PRCA) minkä tahansa erytropoietiinihoidon jälkeen, ei tule hoitaa EPREXillä tai muilla erytropoietiinilla (ks. Kohta 4.4 PRCA).
Hallitsematon verenpaine.
Kaikki autologiseen veren esipohjaistamisohjelmaan liittyvät vasta-aiheet on pidettävä mielessä EPREX-hoitoa saavilla potilailla.
EPREXin käyttö on vasta -aiheista, jos potilaalla on vakavia verisuonisairauksia sepelvaltimo-, perifeerinen valtimo-, kaula- tai aivotasolla, jotka ovat ehdokkaita suureen elektiiviseen ortopediseen leikkaukseen eivätkä ole osa autologista luovutusohjelmaa. Käyttö on myös vasta -aiheista. potilailla, joilla on äskettäin ollut sydäninfarkti tai muita aivoverenkierron komplikaatioita.
Potilaat, jotka ovat ehdolla leikkaukseen ja jotka eivät jostain syystä voi saada riittävää antitromboottista ennaltaehkäisyä.
04.4 Varoitukset ja käyttöön liittyvät varotoimet
Yleistä
Kaikkien epoetiini alfaa saavien potilaiden verenpainetta on seurattava ja seurattava tarpeen mukaan. Epoetiini alfaa tulee käyttää varoen, jos potilaalla on hoitamaton, riittämätön hoito tai vaikea verenpainetauti. Verenpainetta alentava hoito on ehkä aloitettava tai tehostettava. Jos verenpainetta ei saada hallintaan, epoetiini alfa -hoito on lopetettava.
Hypertensiivisiä kriisejä, joihin liittyy enkefalopatiaa ja kohtauksia ja jotka vaativat välitöntä lääkärinhoitoa ja intensiivistä lääketieteellistä hoitoa, on esiintynyt epoetiini alfa -hoidon aikana myös potilailla, joilla on normaali tai matala verenpaine. Erityistä huomiota on kiinnitettävä migreenin kaltaisiin haavaumiin mahdollisena varoitusmerkkinä (ks. Kohta 4.8).
Epoetiini alfaa tulee käyttää varoen potilailla, joilla on epilepsia, joilla on ollut kouristuskohtauksia tai sairauksia, jotka liittyvät alttiuteen kouristuskohtauksiin, kuten keskushermostotulehdukset ja aivometastaasit.
Epoetiini alfaa tulee käyttää varoen potilailla, joilla on krooninen maksan vajaatoiminta Epoetiini alfan turvallisuutta ei ole varmistettu potilailla, joilla on maksan vajaatoiminta.
Verisuonten tromboottisten tapahtumien (VTE) ilmaantuvuutta on havaittu potilailla, jotka ovat saaneet ESA: ta (ks. Kohta 4.8) .Näitä ovat laskimo- ja valtimotromboosi ja embolia (mukaan lukien jotkut kuolemaan johtaneet), kuten syvä laskimotukos, keuhkoembolia, verkkokalvo tromboosi ja sydäninfarkti Lisäksi on raportoitu aivoverenkiertohäiriöitä (mukaan lukien aivoinfarkti, aivoverenvuoto ja ohimenevät iskeemiset kohtaukset).
Näiden laskimotromboembolioiden riskiä on punnittava huolellisesti suhteessa epoetiini alfa -hoidon hyötyyn erityisesti potilailla, joilla on ennestään laskimotromboembolian riskitekijöitä, mukaan lukien liikalihavuus ja aiempi laskimotromboembolia (esim. Syvä laskimotukos, keuhkoembolia ja aivoverisuonitapaukset)
Hemoglobiinipitoisuuksia on seurattava tarkasti kaikilla potilailla, koska tromboembolisten tapahtumien ja kuolemaan johtavien seurausten riski saattaa kasvaa, kun potilaita hoidetaan hemoglobiinipitoisuudella, joka ylittää ilmoitetun pitoisuuden.
Epoetiini alfa -hoidon aikana saattaa esiintyä kohtalaista annoksesta riippuvaa verihiutaleiden määrän nousua, vaikkakin normaalin rajojen sisällä. Tämä ilmiö taantuu hoidon aikana. Lisäksi on raportoitu normaalia korkeampaa trombosytemiaa.Trombosyyttejä suositellaan seuraamaan säännöllisesti ensimmäisen 8 hoitoviikon aikana.
Kaikki mahdolliset anemian syyt (raudanpuute, folaatti- tai B12 -vitamiinin puutos, alumiinimyrkytys, infektio tai tulehdus, verenhukka, hemolyysi ja minkä tahansa alkuperän luuydinfibroosi) on arvioitava ja hoidettava ennen hoidon aloittamista. Epoetiini alfa ja kun päätös tehdään lisäämään annosta. Useimmissa tapauksissa seerumin ferritiiniarvot pienenevät samanaikaisesti hematokriittiarvojen nousun kanssa. Optimaalisen vasteen varmistamiseksi epoetiini alfaan on varmistettava riittävät rautavarastot ja tarvittaessa annettava raudan lisäravinteita ( katso kohta 4.2):
• Kroonista munuaisten vajaatoimintaa sairastaville potilaille raudan lisäystä suositellaan, jos ferritiinipitoisuus on alle 100 ng / ml (alkuainerauta aikuisille 200-300 mg / vrk suun kautta ja lapsille 100-200 mg / vrk suun kautta).
• Syöpäpotilaille raudan lisäystä suositellaan, jos transferriinin kyllästymisarvot ovat alle 20% (alkuainerauta 200 - 300 mg / vrk suun kautta).
• Potilaille, jotka osallistuvat autologiseen luovutusohjelmaan, raudan lisäravinteet (alkuainerauta 200 mg / vrk suun kautta) on annettava muutama viikko ennen autologisen luovutuksen alkamista, jotta saavutetaan suuret rautavarastot. Ennen epoetiini alfa -hoidon aloittamista ja hoidon aikana epoetiini alfa -hoidosta.
• Potilaille, joille on määrätty suuri elektiivinen ortopedinen leikkaus, on annettava rautaa (alkuainerauta 200 mg / vrk suun kautta) epoetiini alfa -hoidon aikana. "Rautalisää ennen epoetiini alfa -hoidon aloittamista riittävän rautavarannon saavuttamiseksi.
Porfyrian alkamista tai pahenemista on havaittu hyvin harvoin epoetiini alfalla hoidetuilla potilailla.
Epoetiini alfaa tulee käyttää varoen potilailla, joilla on porfyria.
Erytropoieesia stimuloivien aineiden (ESA) jäljitettävyyden varmistamiseksi annetun ESA: n kaupallinen nimi on aina rekisteröitävä tai ilmoitettava potilaan sairauskertomuksessa.
Hoidon vaihtaminen EKT: sta toiseen tulee tehdä vain asianmukaisen valvonnan alaisena.
Puhdas punasoluaplasia (PRCA)
Vasta-ainevälitteistä puhdasta punasoluaplasiaa (PRCA) on raportoitu kuukausien ja vuosien jälkeen pääasiassa kroonista munuaisten vajaatoimintaa sairastavilla potilailla, joita on hoidettu ihon alle annettavalla epoetiinilla.
Tapauksia on raportoitu myös hepatiitti C -potilailla, joita hoidettiin interferonilla ja ribaviriinilla, kun niitä annettiin yhdessä ESA: n kanssa. Epoetiini alfaa ei ole hyväksytty hepatiitti C: hen liittyvän anemian hoitoon.
Potilaille, joiden teho heikkenee äkillisesti, määritettynä hemoglobiiniarvojen laskuna (1-2 g / dl kuukaudessa) ja joilla on lisääntynyt verensiirtotarve, on suoritettava retikulosyyttien lukumäärä ja tiedettävä syyt, jotka estävät vaste (kuten raudan, folaatin ja B12 -vitamiinin puutos, alumiinimyrkytys, infektio tai tulehdus, verenhukka, hemolyysi ja minkä tahansa alkuperän luuydinfibroosi).
Jos hemoglobiiniarvot laskevat suhteettomasti ja kehittyy vaikea anemia, johon liittyy alhainen retikulosyyttimäärä, sen pitäisi johtaa epoetiini alfa -hoidon lopettamiseen ja testiin erytropoietiinivasta-aineiden esiintymisen varalta.
Myös luuydintutkimusta PRCA -diagnoosin saamiseksi on harkittava.
Ristireaktion riskin vuoksi muita ESA -lääkkeitä ei saa aloittaa.
Oireisen anemian hoito kroonista munuaisten vajaatoimintaa sairastavilla aikuis- ja lapsipotilailla:
Kroonista munuaisten vajaatoimintaa sairastavilla potilailla, jotka saavat epoetiini alfaa, hemoglobiinitasot on mitattava säännöllisesti, kunnes vakaa taso saavutetaan, ja mitattava sitten säännöllisesti sen jälkeen.
Potilailla, joilla on krooninen munuaisten vajaatoiminta verenpaineen nousun riskin pienentämiseksi, hemoglobiinin tulisi nousta noin 1 g / dl / kk (0,62 mmol / l) eikä se saa ylittää 2 g / dl / kk (1,25 mmol / l).
Potilailla, joilla on krooninen munuaisten vajaatoiminta, ylläpitävä hemoglobiinipitoisuus ei saa ylittää kohdassa 4.2 ilmoitettua hemoglobiinipitoisuuden vaihteluvälin maksimiarvoa. Kliinisissä tutkimuksissa on havaittu lisääntynyttä kuolleisuuden ja vakavien sydän- ja verisuonitapahtumien riskiä, kun erytropoieesiä stimuloivia aineita (ESA) annetaan hemoglobiinipitoisuuden saavuttamiseksi yli 12 g / dl (7,5 mmol / ml).
Kontrolloidut kliiniset tutkimukset eivät ole osoittaneet merkittävää hyötyä epoetiinien antamisesta, kun hemoglobiinipitoisuus ylittää anemian oireiden hallitsemiseksi ja verensiirtojen välttämiseksi tarvittavan tason.
Varovaisuutta on noudatettava EPREX -annoksen suurentamisessa potilailla, joilla on krooninen munuaisten vajaatoiminta, koska suuret kumulatiiviset epoetiiniannokset voivat lisätä kuolleisuuden, vakavien sydän- ja aivoverisuonitapahtumien riskiä. Potilaille, joilla on huono hemoglobiinivaste. tähän huonoon vasteeseen on löydettävä syy (ks. kohdat 4.2 ja 5.1).
Kroonista munuaisten vajaatoimintaa sairastavia potilaita, joita hoidetaan ihonalaisella epoetiini alfalla, on seurattava säännöllisesti tehon heikkenemisen varalta, mikä tarkoittaa, että epoetiini alfaa ei ole tai vaste on heikentynyt potilailla, jotka ovat aikaisemmin saaneet hoidon. Tälle ilmiölle on ominaista jatkuva hemoglobiiniarvojen lasku verrattuna epoetiini alfa -annoksen suurentamiseen (ks. Kohta 4.8).
Jotkut potilaat, joita hoidetaan epoetiini alfaa pitemmillä annosvälillä (useammin kuin kerran viikossa), eivät ehkä ylläpitää riittävää hemoglobiinitasoa (ks. Kohta 5.1) ja saattavat vaatia annoksen suurentamista. Hemoglobiinipitoisuuksia on seurattava säännöllisesti.
Verisuonitukosten tromboosia on esiintynyt hemodialyysipotilailla, erityisesti potilailla, joilla on taipumus hypotensioon ja joilla on arteriovenoosisten fistulien komplikaatioita (esim. Ahtauma, aneurysmat jne.) .Näillä potilailla suositellaan verisuonten pääsyn ennaltaehkäisevää valvontaa ja tromboosin ennaltaehkäisyä esim. asetyylisalisyylihappoa.
Hyperkalemiaa on havaittu yksittäistapauksissa, vaikka syy -yhteyttä ei ole osoitettu. Seerumin elektrolyyttejä on seurattava potilailla, joilla on krooninen munuaisten vajaatoiminta. Jos seerumin kaliumpitoisuus on kohonnut (tai nouseva), hyperkalemian asianmukaisen hoidon lisäksi on harkittava epoetiini alfa -hoidon lopettamista, kunnes seerumin kaliumpitoisuus on korjattu.
Usein hemodialyysin aikana hepariiniannosta on lisättävä hematokriittiarvon nousun vuoksi. Jos hepariiniannosten säätäminen ei ole optimaalista, dialysaattori voi tukkeutua. Tähän mennessä saatavilla olevien tietojen perusteella anemian korjaaminen epoetiini alfalla aikuispotilailla, joilla on krooninen munuaisten vajaatoiminta ja jotka eivät ole vielä dialyysissä, ei nopeuta munuaisten vajaatoiminnan etenemistä.
Potilaiden hoito, joilla on kemoterapian aiheuttama anemia
Syöpäpotilaiden, jotka saavat epoetiini alfaa, hemoglobiinitasot on mitattava säännöllisesti, kunnes vakaa taso saavutetaan, ja sitten mitattava määräajoin sen jälkeen.
Erytropoietiinit ovat kasvutekijöitä, jotka olennaisesti stimuloivat punasolujen tuotantoa. Erytropoietiinireseptoreita voidaan ekspressoida useiden syöpäsolujen pinnalla. Kuten kaikkien kasvutekijöiden kohdalla, on teoreettista huolta siitä, että erytropoietiinit voivat stimuloida kasvaimen kasvua.
Joissakin kontrolloiduissa kliinisissä tutkimuksissa erytropoietiinien ei ole osoitettu parantavan kokonaiseloonjäämistä tai vähentävän kasvaimen etenemisen riskiä potilailla, joilla on kasvaimeen liittyvä anemia.
Kontrolloiduissa kliinisissä tutkimuksissa epoetiini alfan ja muiden erytropoieesiä stimuloivien aineiden (ESA) käyttö on osoittanut:
• heikentynyt paikallisalueen valvonta potilailla, joilla on pitkälle edennyt pään ja kaulan syöpä ja jotka saavat sädehoitoa, kun hemoglobiinipitoisuus on yli 14 g / dl (8,7 mmol / l);
• vähentynyt kokonaiseloonjääminen ja lisääntynyt kuolleisuus johtuen taudin etenemisestä 4 kuukauden kohdalla potilailla, joilla on metastasoitunut rintasyöpä ja jotka saavat solunsalpaajahoitoa annettaessa hemoglobiinitasoja 12-14 g / dl (7,5-8,7 mmol / l);
• lisääntynyt kuolleisuusriski, kun sitä annetaan, kun hemoglobiinitaso on 12 g / dl (7,5 mmol / l); potilailla, joilla on aktiivinen neoplasia ja jotka eivät saa kemo- ja / tai sädehoitoa. Hoitoa erytropoieesia stimuloivilla aineilla (ESA) ei ole tarkoitettu tähän potilasryhmään.
Edellä esitetyn perusteella joissakin kliinisissä tilanteissa anemian hoidossa syöpäpotilailla tulisi suosia verensiirtoa. Päätös yhdistelmä-erytropoietiinin antamisesta tulee perustaa hyöty-riskiarviointiin, johon potilas osallistuu yksilöllisesti. Arvioinnissa on otettava huomioon syövän tyyppi ja sen eteneminen. anemian aste; elinajanodote, ympäristö, jossa potilasta hoidetaan; potilaan mieltymykset (ks. kohta 5.1).
Solunsalpaajahoitoa saavilla syöpäpotilailla 2-3 viikon välein annostelun ja ESA: n aiheuttamien punasolujen esiintymisen välillä on otettava huolellisesti huomioon arvioitaessa epoetiini alfa -hoidon tarkoituksenmukaisuutta (verensiirtoriski).
Aikuispotilaat, jotka ovat ehdokkaita kirurgisiin toimenpiteisiin, jotka ovat osa autologisia verenluovutusohjelmia
Kaikkia varoituksia ja erityisiä varotoimia, jotka liittyvät autologiseen verenluovutusohjelmaan, erityisesti rutiininomaiseen tilavuuden korvaamiseen, on noudatettava.
Potilaat, jotka ovat ehdolla suureen valinnaiseen ortopediseen leikkaukseen
Hyviä verenhallintatapoja tulee aina noudattaa ennen leikkausta.
Potilaita, jotka ovat ehdokkaita valinnaiseen suureen ortopediseen leikkaukseen, tulee saada "riittävä antitromboottinen profylaksia, koska tromboottisia ja verisuonitapahtumia voi esiintyä leikkauksessa olevilla potilailla, erityisesti niillä, joilla on taustalla sydän- ja verisuonitauteja. Lisäksi on noudatettava erityisiä varotoimia potilailla, joilla on taipumus syvien laskimotromboosien (DVT) kehittymistä. Lisäksi potilaita, joiden "lähtötason hemoglobiini> 13 g / dl, ei voida sulkea pois mahdollisuutta, että epoetiini alfa -hoitoon voi liittyä kohonnut tromboottisten / verisuonitapahtumien riski." . Siksi epoetiini alfaa ei tule käyttää potilailla, joiden lähtötason hemoglobiini on> 13 g / dl.
04.5 Yhteisvaikutukset muiden lääkevalmisteiden kanssa sekä muut yhteisvaikutukset
Ei ole näyttöä siitä, että epoetiini alfa -hoito muuttaisi muiden lääkkeiden aineenvaihduntaa. Lääkkeet, jotka vähentävät erytropoieesia, voivat heikentää epoetiini alfavastetta.
Koska syklosporiini sitoutuu punasoluihin, voi olla vuorovaikutusta tämän lääkkeen kanssa.
Samanaikaisen annon yhteydessä siklosporiinin pitoisuuksia veressä on seurattava ja annosta säädettävä hematokriitin nousun mukaan.
G-CSF: n, GM-CSF: n ja epoetiini alfan välillä ei ole in vitro -interaktioita hematologisen erilaistumisen tai lisääntymisen suhteen in vitro kasvaimen biopsianäytteistä.
Aikuisilla naispotilailla, joilla oli metastasoitunut rintasyöpä, 40000 IU / ml epoetiini alfaa annettiin ihon alle samanaikaisesti trastutsumabin 6 mg / kg kanssa, eikä sillä ollut vaikutusta trastutsumabin farmakokinetiikkaan.
04.6 Raskaus ja imetys
Raskaus
Raskaana oleville naisille ei ole tehty riittäviä ja hyvin kontrolloituja tutkimuksia. Eläinkokeet ovat osoittaneet lisääntymistoksisuutta (ks. Kohta 5.3). Näin ollen epoetiini alfaa tulee käyttää raskauden aikana vain, jos odotettu hyöty on suurempi kuin mahdollinen riski sikiölle. Epoetiini alfan käyttöä ei suositella raskaana oleville naisille, jotka ovat ehdolla leikkaukseen ja jotka osallistuvat autologiseen verenluovutusohjelmaan.
Ruokinta-aika
Ei tiedetä, erittyykö eksogeeninen epoetiini alfa äidinmaitoon. Epoetiini alfaa tulee käyttää varoen imettäville naisille. Päätös imetyksen jatkamisesta / lopettamisesta tai epoetiini alfa -hoidon jatkamisesta / lopettamisesta tulee tehdä ottaen huomioon imetyksen hyödyt vauva ja epoetiini alfa -hoidon hyödyt naiselle.
Epoetiini alfan käyttöä ei suositella imettäville naisille, jotka ovat ehdolla leikkaukseen ja jotka osallistuvat autologiseen verenluovutusohjelmaan.
Hedelmällisyys
Ei ole tutkimuksia, joissa arvioitiin epoetiini alfan mahdollista vaikutusta miesten tai naisten hedelmällisyyteen.
04.7 Vaikutus ajokykyyn ja koneiden käyttökykyyn
Tutkimuksia ajokyvystä tai koneiden käyttökyvystä ei ole tehty.
04.8 Haittavaikutukset
Yhteenveto turvallisuusprofiilista
Yleisin lääkkeen haittavaikutus epoetiini alfa -hoidon aikana on annoksesta riippuva verenpaineen nousu tai aiemman verenpaineen paheneminen.
Verenpaineen kehitystä on suositeltavaa seurata erityisesti hoidon alussa (ks. Kohta 4.4).
Epoetiini alfan kliinisissä tutkimuksissa esiintyneet yleisimmät haittavaikutukset ovat ripuli, pahoinvointi, oksentelu, kuume ja päänsärky. Flunssan kaltaisia oireita voi esiintyä pääasiassa hoidon alussa.
Hengitysteiden tukkoisuutta, mukaan lukien ylempien hengitysteiden tukkoisuutta, nenän tukkoisuutta ja nenä-nielutulehdusta, on raportoitu annosvälin laajentamistutkimuksissa aikuispotilailla, joilla on munuaisten vajaatoiminta ja jotka eivät ole vielä dialyysihoidossa.
Verisuonten tromboottisten tapahtumien (TVE) ilmaantuvuutta on havaittu potilailla, jotka ovat saaneet ESA: ta (ks. Kohta 4.4).
Taulukko haittavaikutuksista
Kaikista 3262 potilaasta 23 satunnaistetussa, kaksoissokkoutetussa, lumelääkkeellä tai tavanomaisella hoidolla kontrolloidussa kliinisessä tutkimuksessa EPREXin yleistä turvallisuusprofiilia arvioitiin 1 922 aneemisella potilaalla. Neljään kroonista munuaisten vajaatoimintaa koskevaan tutkimukseen osallistui 228 epoetiini alfalla hoidettua ICR-potilasta (2 tutkimusta esidialyysistä [N = 131 ICR-altistettua potilasta] ja 2 dialyysihoitoa [N = 97 ICR-potilaat]); 1404 syöpäpotilasta altistui 16 tutkimuksessa kemoterapian aiheuttamasta anemiasta; 147 potilasta, jotka altistuivat kahdessa tutkimuksessa autologisen veren luovutusta varten ja 213 potilasta altistui 1 tutkimuksessa perioperatiivisen ajanjakson aikana. Haittavaikutuksia raportoitiin ≥ 1%: lla epoetiini alfa näissä kliinisissä tutkimuksissa on esitetty alla olevassa taulukossa.
Seuraavat määritelmät koskevat eri esiintymistiheyksiä: hyvin yleinen (≥ 1/10), yleinen (≥ 1/100 -
Valittujen haittavaikutusten kuvaus
Yliherkkyysreaktioita, mukaan lukien ihottuma (mukaan lukien nokkosihottuma), anafylaktisia reaktioita ja angioedeemaa on raportoitu.
Hypertensiivisiä kriisejä, joihin liittyy enkefalopatiaa ja kohtauksia, jotka vaativat välitöntä lääkärinhoitoa ja intensiivistä lääketieteellistä hoitoa, on esiintynyt epoetiini alfa -hoidon aikana myös potilailla, joilla on normaali tai matala verenpaine. Erityistä huomiota on kiinnitettävä migreenin kaltaisiin haavaumiin mahdollisena varoitusmerkkinä (ks. Kohta 4.4).
Vasta-ainevälitteistä puhdasta punasoluaplasiaa on raportoitu hyvin harvoin vuonna
Pediatriset potilaat, joilla on krooninen munuaisten vajaatoiminta hemodialyysissä
Hemodialyysihoitoa saavien kroonisten munuaisten vajaatoimintaa sairastavien lasten altistus kliinisissä tutkimuksissa ja markkinoille tulon jälkeen on rajallinen. Tässä potilasryhmässä ei raportoitu mitään erityisiä haittavaikutuksia lapsipopulaatiosta, joita ei ole mainittu yllä olevassa taulukossa, tai perussairaukselle tyypillisiä haittavaikutuksia.
Epäillyistä haittavaikutuksista ilmoittaminen
Ilmoittaminen epäillyistä haittavaikutuksista, jotka ilmenevät lääkkeen myyntiluvan myöntämisen jälkeen, on tärkeää, koska sen avulla voidaan jatkuvasti seurata lääkkeen hyöty -haitta -suhdetta. Terveydenhuollon ammattilaisia pyydetään ilmoittamaan kaikista epäillyistä haittavaikutuksista kansallisen ilmoitusjärjestelmän kautta. "Osoite http: //www.agenziafarmaco.gov.it/it/responsabili.
04.9 Yliannostus
Epoetiini alfan terapeuttinen marginaali on erittäin laaja. Epoetiini alfan yliannostus voi aiheuttaa vaikutuksia, jotka jatkavat hormonin farmakologisia vaikutuksia.Flebotomia voidaan suorittaa, jos havaitaan liian korkeat hemoglobiinitasot.
Lisätukea on annettava potilaan tilan edellyttämällä tavalla.
05.0 FARMAKOLOGISET OMINAISUUDET
05.1 Farmakodynaamiset ominaisuudet
Farmakoterapeuttinen ryhmä: anemia, ATC-koodi: B03XA01.
Toimintamekanismi
Erytropoietiini (EPO) on glykoproteiinihormoni, jonka pääasiassa tuottaa munuaiset vasteena hypoksialle, ja se on punasolujen (RBC) tuotannon keskeinen säätelijä. "EPO on mukana kaikissa erytroidisen kehityksen vaiheissa ja sillä on pääasiallinen vaikutus punasolujen esiasteiden tasolla. Kun EPO sitoutuu reseptoriinsa solun pinnalla, se aktivoi signaalinsiirtoreitit, jotka häiritsevät" apoptoosia ja stimuloivat punasolujen lisääntymistä. Rekombinantti ihmisen erytropoietiinilla (epoetiini alfa), joka ilmaistaan kiinalaisen hamsterin munasarjasoluissa, on 165 aminohapon sekvenssi, joka on identtinen ihmisen virtsan EPO: n kanssa; kaksi ovat erottamattomia toiminnallisten määritysten perusteella. Erytropoietiinin näennäinen molekyylipaino on 32 000 - 40 000 daltonit.
Erytropoietiini on kasvutekijä, joka stimuloi ensisijaisesti punasolujen tuotantoa.Erytropoietiinireseptoreita voidaan ekspressoida useiden syöpäsolujen pinnalla.
Farmakodynaamiset vaikutukset
Terveitä vapaaehtoisia
Yksittäisten epoetiini alfa -annosten jälkeen (20000-160 000 IU ihon alle) havaittiin annoksesta riippuva vaste tutkituille farmakodynaamisille markkereille, mukaan lukien: retikulosyytit, punasolut ja hemoglobiini. Määritelty pitoisuus-ajallinen profiili havaittiin huipulla ja palautui lähtötilanteeseen retikulosyyttien prosenttiosuuden muutosten osalta. Punasolujen ja hemoglobiinin profiilia havaittiin vähemmän. Yleensä kaikki farmakodynaamiset markkerit kasvoivat lineaarisesti annosten saavuttaessa maksimaalisen vasteen suurimmilla annostasoilla.
Muissa farmakodynaamisissa tutkimuksissa tutkittiin 40 000 IU kerran viikossa verrattuna 150 IU / kg 3 kertaa viikossa. Pitoisuus-aikaprofiilien eroista huolimatta farmakodynaaminen vaste (mitattuna prosentuaalisin muutoksin retikulosyyteissä, hemoglobiinissa ja punasolujen kokonaismäärässä) oli samanlainen näillä hoito-ohjelmilla. Lisätutkimuksissa verrattiin 40000 IU epoetiini alfa -hoitoa kerran viikossa kahden viikon välein annoksiin, jotka vaihtelivat 80000 - 120 000 IU ihon alle. Kaiken kaikkiaan näiden terveillä henkilöillä tehtyjen farmakodynaamisten tutkimusten tulosten perusteella 40 000 IU kerran viikossa annosteluohjelma näyttää olevan tehokkaampi punasolujen tuotannossa kuin kahden viikon välein, vaikka retikulosyyttituotanto on samankaltainen. Kerran viikossa ja kahden viikon välein.
Krooninen munuaisten vajaatoiminta
Epoetiini alfan on osoitettu stimuloivan erytropoieesia aneemisilla potilailla, joilla on krooninen munuaisten vajaatoiminta, mukaan lukien dialyysi- ja dialyysipotilaat. Ensimmäinen todiste epoetiini alfa -vasteesta on retikulosyyttien määrän lisääntyminen 10 päivän kuluessa, mitä seuraa punasolujen, hemoglobiinin ja hematokriitin määrän nousu, yleensä 2-6 viikon kuluessa. Hemoglobiinivaste vaihtelee potilaiden välillä, ja raudan kertyminen ja samanaikaiset lääketieteelliset ongelmat voivat vaikuttaa siihen.
Kemoterapian aiheuttama anemia
3 kertaa viikossa tai kerran viikossa annetun epoetiini alfan on osoitettu lisäävän hemoglobiinia ja vähentävän verensiirtojen tarvetta ensimmäisen hoitokuukauden jälkeen anemiaa sairastavilla syöpäpotilailla.
Tutkimuksessa, jossa verrattiin 150 IU / kg annoksia 3 kertaa viikossa ja 40000 IU kerran viikossa terveillä koehenkilöillä ja aneemisilla syöpäpotilailla, retikulosyyttien, hemoglobiinin ja punasolujen kokonaismäärän prosentuaalisten muutosten ajalliset profiilit olivat samankaltaisia kaksi annostusohjelmaa terveillä ja aneemisilla syöpäpotilailla. Vastaavien farmakodynaamisten parametrien AUC -arvot olivat samankaltaiset 150 IU / kg 3 kertaa viikossa ja 40 000 IU kerran viikossa annostusohjelmilla sekä terveillä että aneemisilla syöpäpotilailla.
Aikuiset kirurgiset potilaat autologisessa luovutusohjelmassa
Epoetiini alfan on osoitettu stimuloivan punasolujen tuotantoa autologisen veren keräämisen lisäämiseksi ja hemoglobiinipudotuksen rajoittamiseksi aikuisilla potilailla, joille on määrä tehdä suuri elektiivinen leikkaus ja joille ei odoteta täydellistä perioperatiivista esivarastointia. Suurimmat vaikutukset havaittiin potilailla, joilla oli alhainen hemoglobiinipitoisuus (≤ 13 g / dl).
Suurten elektiivisten ortopedisten leikkausten hoitoon tarkoitettujen aikuispotilaiden hoito
Epoetiini alfan on osoitettu vähentävän allogeenisten verensiirtojen riskiä ja nopeuttavan punasolujen palautumista (kohonnut hemoglobiinipitoisuus, hematokriitti ja retikulosyyttimäärä) potilailla, joille on suunniteltu suuri elektiivinen ortopedinen leikkaus ja joiden hemoglobiinin esikäsittely on> 10 - ≤ 13 g / dl.
Kliininen teho ja turvallisuus
Krooninen munuaisten vajaatoiminta
Epoetiini alfaa on arvioitu kliinisissä tutkimuksissa aneemisilla aikuispotilailla, joilla on CRF, mukaan lukien hemodialyysipotilaat ja esidialyysipotilaat, anemian hoitamiseksi ja hematokriitin säilyttämiseksi pitoisuusalueella 30-36%.
Kliinisissä tutkimuksissa aloitusannoksilla 50-150 IU / kg kolme kertaa viikossa noin 95% kaikista potilaista vastasi kliinisesti merkittävään hematokriitin nousuun. Noin kahden kuukauden hoidon jälkeen lähes kaikki potilaat saivat verensiirron. hematokriittitavoite saavutettiin, ylläpitoannos räätälöitiin kullekin potilaalle.
Kolme suurta kliinistä tutkimusta aikuisilla dialyysipotilailla keskimääräinen ylläpitoannos, joka tarvittiin hematokriitin ylläpitämiseksi 30 ja 36% välillä, oli noin 75 IU / kg 3 kertaa viikossa.
Kaksoissokkoutetussa, monikeskustutkimuksessa, lumekontrolloidussa tutkimuksessa hemodialyysipotilaiden CRF-potilaiden elämänlaadusta, kliinisesti ja tilastollisesti merkitsevä paraneminen osoitettiin epoetiini alfa -hoitoa saaneilla potilailla lumelääkeryhmään verrattuna, mitaten väsymystä, fyysisiä oireita, ihmissuhteita ja masennus (munuaistautikysely) kuuden kuukauden hoidon jälkeen. Epoetiini alfa -ryhmän potilaat osallistui avoimeen jatkotutkimukseen, joka osoitti, että heidän elämänlaadunsa parani vielä 12 kuukautta.
Aikuiset munuaisten vajaatoimintaa sairastavat potilaat, jotka eivät ole vielä dialyysihoidossa
Kliinisissä tutkimuksissa CRF -potilailla, jotka eivät olleet dialyysihoitoa saaneet epoetiini alfaa, hoidon keskimääräinen kesto oli lähes viisi kuukautta. Nämä potilaat vastasivat epoetiini alfa -hoitoon samalla tavalla kuin dialyysipotilailla. CRF -potilaat, jotka eivät olleet dialyysissä, osoittivat annosriippuvuutta ja tasaista hematokriitin nousua, kun epoetiini alfaa annettiin sekä suonensisäisesti että ihonalaisesti. Samanlaisia hematokriitin kasvunopeuksia havaittiin, kun epoetiini alfaa annettiin molemmille. Lisäksi epoetiini alfa -annokset 75--150 IU / kg viikossa on osoitettu ylläpitävän hematokriitti 36-38%: ssa jopa kuuden kuukauden ajan.
Kahdessa pidennetyssä EPREX -annosvälitutkimuksessa (3 kertaa viikossa, kerran viikossa, kerran kahdessa viikossa ja joka neljäs viikko) jotkut potilaat, joilla oli pidempi annostusväli, eivät ylläpitäneet riittävää hemoglobiinitasoa ja täyttivät vakiintuneet vieroituskriteerit (0% kerran viikossa, 3,7% 2 viikon välein ja 3,3% 4 viikon välein).
Prospektiivisessa satunnaistetussa tutkimuksessa (CHOIR) arvioitiin 1432 aneemista potilasta, joilla oli krooninen munuaisten vajaatoiminta ja jotka eivät olleet dialyysissä. Potilaille annettiin epoetiini alfa -hoitoa, jonka tavoitteena oli ylläpitää hemoglobiinitaso 13,5 g / dl (suositellun hemoglobiinitason yläpuolella) tai 11,3 g / dl. Suuri sydän- ja verisuonitapahtuma (kuolema, sydäninfarkti, aivohalvaus tai sairaalahoito kongestiivisen sydämen vajaatoiminnan vuoksi) , esiintyi 125 (18%) 715 potilaan joukossa korkeammassa hemoglobiiniryhmässä kuin 97 (14%) 717 potilaan joukossa alemman hemoglobiinin ryhmässä (riskin prosenttiosuus [HR] 1,3, 95%CI: 1,0, 1,7, p = 0,03).
Kroonista munuaisten vajaatoimintaa sairastavilla potilailla (dialyysi- ja ei-dialyysipotilailla, diabeetikoilla ja ei-diabeetikoilla) suoritettiin kumulatiiviset retrospektiiviset analyysit kliinisistä ESA-tutkimuksista. Potilaan diabetes- tai dialyysitilasta riippumatta havaittiin suuntaus lisääntyvään arvioituun riskiin kaikista syistä johtuvaan kuolleisuuteen, sydän- ja aivoverisuonitapahtumiin, jotka liittyivät suurempiin kumulatiivisiin ESA -annoksiin (ks. Kohdat 4.2 ja 4.4).
Kemoterapian aiheuttaman anemian hoito
Epoetiini alfaa on arvioitu kliinisissä tutkimuksissa aikuisilla aneemisilla syöpäpotilailla, joilla on imusolmukkeita ja kiinteitä kasvaimia, sekä potilailla, jotka ovat saaneet erilaisia kemoterapiahoitoja, mukaan lukien platina- ja ei-platinahoitoja. Näissä tutkimuksissa 3 kertaa viikossa ja kerran viikossa annetun epoetiini alfan osoitettiin lisäävän hemoglobiinia ja vähentävän verensiirtojen tarvetta ensimmäisen hoitokuukauden jälkeen aneemisilla syöpäpotilailla. Joissakin tutkimuksissa kaksoissokkoutettua vaihetta seurasi avoin vaihe, jonka aikana kaikki potilaat saivat epoetiini alfaa ja vaikutuksen säilymistä.
Käytettävissä olevat tiedot viittaavat siihen, että potilaat, joilla on hematologisia maligniteetteja ja kiinteitä kasvaimia, reagoivat vastaavasti epoetiini alfa -hoitoon ja että potilaat, joilla on luuytimen kasvainsuodatus tai ilman sitä, vastaavat vastaavasti epoetiini alfa -hoitoa. Kemoterapiatutkimuksissa vertailukelpoinen kemoterapian intensiteetti epoetiini alfa- ja lumelääkeryhmissä osoitti samanlaisen alueen neutrofiilien aikakäyrän alla epoetiini alfaa ja lumelääkettä saaneilla potilailla sekä vastaava osuus potilaista hoidetuissa ryhmissä. epoetiini alfa ja lumelääkeryhmät, joiden absoluuttinen neutrofiilimäärä laski alle 1000 ja 500 solua / ml
Prospektiivisessa, kaksoissokkoutetussa, satunnaistetussa ja lumekontrolloidussa tutkimuksessa, johon osallistui 375 aneemista potilasta, joilla oli ei-myelooisia kasvaimia, jotka saivat ei-platinapohjaista solunsalpaajahoitoa, anemiaan liittyvien oireiden (kuten astenian, väsymyksen ja verenpaineen alenemisen) merkittävä väheneminen aktiivisuus), mitattuna seuraavilla arviointiasteikoilla: Syöpäterapian anemian toiminnallinen arviointi (FACT-An) yleinen asteikko; FACT-väsymysasteikko ja syövän lineaarinen analogiasteikko (CLAS).
Kahdessa muussa satunnaistetussa ja lumelääkekontrolloidussa tutkimuksessa, jotka tehtiin pienemmällä määrällä potilaita, ei havaittu parannusta elämänlaatuparametreissa EORTC-QLQ-C30- ja CLAS-asteikon mukaan.
Selviytymistä ja kasvaimen etenemistä tutkittiin viidessä suuressa kontrolloidussa tutkimuksessa, joihin osallistui yhteensä 2833 potilasta, mukaan lukien neljä kaksoissokkoutettua lumekontrolloitua ja yksi avoin tutkimus. Tutkimuksiin osallistui potilaita, jotka saivat sekä kemoterapiahoitoa (kaksi tutkimusta) että potilaita, joille ESA: n käyttöä ei ollut osoitettu: aneemiset syöpäpotilaat, jotka eivät saa solunsalpaajahoitoa, ja potilaat, joilla on pään ja kaulan syöpä, jotka saavat sädehoitoa. Haluttu sädehoidon pitoisuustaso. Hemoglobiini kahdessa Tutkimukset olivat> 13 g / dl; muissa kolmessa tutkimuksessa se oli 12-14 g / dl. Avoimessa tutkimuksessa eloonjäämisessä ei ollut eroa rekombinantilla ihmisen erytropoietiinilla hoidettujen potilaiden ja kontrollien välillä. Neljässä kontrolloidussa tutkimuksessa"riskisuhde kokonaiseloonjäämisen osalta se vaihteli 1,25 - 2,47 kontrollien hyväksi. Nämä tutkimukset osoittivat selittämättömän tilastollisesti merkitsevän kuolleisuuden lisääntymisen syöpäanemiapotilailla, joita hoidettiin yhdistelmä -DNA -tekniikalla tehdyllä ihmisen erytropoietiinilla verrattuna kontrolleihin. Ihmisen yhdistelmä -erytropoietiinia saaneiden potilaiden eloonjäämisen kokonaistulosta verrokkiryhmään verrattuna ei voitu tyydyttävästi selittää tromboosin ja siihen liittyvien komplikaatioiden ilmaantuvuuserolla.
Potilaatason analyysi tehtiin myös yli 13 900 syöpäpotilaalle (kemo-, radio-, radiokemio- tai ei-hoidossa), jotka osallistuivat 53 kontrolloituun kliiniseen tutkimukseen eri epoetiineilla. Kokonaiseloonjäämistietojen meta-analyysi osoitti täsmällisen riskisuhteen arvioksi 1,06 kontrollien hyväksi (95%: n luottamusväli: 1,00, 1,12; 53 tutkimusta ja 13933 potilasta), kun taas kemoterapiaa saavien syöpäpotilaiden kokonaiselossaolo riskisuhde, se oli 1,04 (95%: n luottamusväli: 0,97, 1,11; 38 tutkimusta ja 10441 potilasta). Lisäksi meta-analyysit osoittavat johdonmukaisesti, että tromboembolisten tapahtumien suhteellinen riski on merkittävästi lisääntynyt syöpäpotilailla, jotka saavat yhdistelmä-DNA-tekniikalla saatua ihmisen erytropoietiinia (ks. Kohta 4.4).
Autologinen predonation -ohjelma
Epoetiini alfan vaikutusta autologisen verenluovutuksen helpottamiseen potilailla, joilla on alhainen hematokriitti (≤ 39% ja ilman selvää raudanpuutteen aiheuttamaa anemiaa), jotka on suunniteltu suureen ortopediseen leikkaukseen, arvioitiin kaksinkertaisessa lumekontrolloidussa tutkimuksessa. Sokea 204 potilaalla ja yhdellä -sokea lumekontrolloitu tutkimus 55 potilaalla.
Kaksoissokkotutkimuksessa potilaita hoidettiin epoetiini alfa 600 IU / kg tai lumelääkkeellä laskimoon kerran vuorokaudessa 3 tai 4 päivän välein 3 viikon ajan (yhteensä 6 annosta). Epoetiini alfalla hoidetut potilaat pystyivät keskimäärin tallentamaan merkittävästi enemmän veriyksiköitä (4,5 yksikköä) kuin lumelääkettä saaneet potilaat (3,0 yksikköä).
Yksisokkoutetussa tutkimuksessa potilaita hoidettiin 300 IU / kg tai 600 IU / kg epoetiini alfaa tai lumelääkettä laskimoon kerran vuorokaudessa 3 tai 4 päivän välein 3 viikon ajan (yhteensä 6 annosta). Epoetiini alfalla hoidetut potilaat pystyivät myös tallentamaan merkittäviä lisäyksiköitä verta (epoetiini alfa 300 IU / kg = 4,4 yksikköä; epoetiini alfa 600 IU / kg = 4,7 yksikköä) verrattuna lumelääkettä saaneisiin potilaisiin (2,9 yksikköä).
Epoetiini alfa -hoito pienensi allogeenisen veren altistumisen riskiä 50% verrattuna potilaisiin, joita ei hoidettu epoetiini alfalla.
Suuri elektiivinen ortopedinen kirurgia
Epoetiini alfan (300 IU / kg tai 100 IU / kg) vaikutusta altistukseen allogeenisille verensiirroille arvioitiin kaksoissokkoutetussa, lumekontrolloidussa kliinisessä tutkimuksessa, jossa oli rautapuutteisia aikuispotilaita, jotka odottivat leikkausta. Epoetiini alfaa annettiin ihon alle 10 päivän ajan ennen leikkausta, leikkauspäivänä ja neljä päivää leikkauksen jälkeen. Potilaat luokiteltiin lähtötilanteen hemoglobiinin mukaan (≤ 10 g / dl,> 10 - ≤ 13 g / dl ja> 13 g / dl).
Epoetiini alfa 300 IU / kg pienensi merkittävästi allogeenisten verensiirtojen riskiä potilailla, joiden hemoglobiini oli ennen hoitoa> 10 - ≤ 13 g / dl. 16% potilaista, jotka saivat epoetiini alfaa 300 IU / kg, 23% epoetiini alfaa 100 IU / kg ja 45% lumelääkettä, tarvitsi verensiirron.
Avoimessa rinnakkaistutkimuksessa aikuisilla, joilla oli raudanpuute, hemoglobiini oli ≥ 10-≤ 13 g / dl ja jotka saivat ortopedistä lonkka- tai polvileikkausta, verrattiin epoetiini alfa 300 IU / kg -hoitoa ihon alle päivittäin 10 päivän ajan ennen leikkausta. leikkauspäivänä ja neljä päivää leikkauksen jälkeen, kun hoidetaan epoetiini alfa 600 IU / kg ihon alle kerran viikossa 3 viikon ajan ennen leikkausta ja leikkauspäivänä.
Esikäsittelystä ennen leikkausta hemoglobiinin keskimääräinen nousu viikoittain 600 IU / kg (1,44 g / dl) -ryhmässä oli kaksinkertainen verrattuna 300 IU / kg päivittäiseen ryhmään (0,73 g / dl). Keskimääräiset hemoglobiinitasot olivat samanlaiset molemmissa hoitoryhmissä koko leikkauksen jälkeisen ajan.
Molemmissa hoitoryhmissä havaittu erytropoieettinen vaste johti samanlaisiin verensiirtonopeuksiin (16% ryhmässä 600 IU / kg / viikko ja 20% ryhmässä 300 IU / kg / päivä)
Pediatriset potilaat
Krooninen munuaisten vajaatoiminta
Epoetiini alfaa arvioitiin avoimessa, ei-satunnaistetussa, avoimessa annosalueella, 52 viikon kliinisessä tutkimuksessa hemodialyysipotilailla. Tutkimukseen osallistuneiden potilaiden keski -ikä oli 11,6 vuotta (vaihteluväli 0,5 - 20,1 vuotta).
Epoetiini alfaa annettiin 75 IU / kg / viikko laskimoon 2 tai 3 jaettuna dialyysin jälkeen, titrattiin 75 IU / kg / viikko 4 viikon välein (enintään 300 IU / kg / viikko), saavuttaa hemoglobiinin nousu 1 g / dl / kk. Keskimääräinen aika määränpäähän oli 11 viikkoa, mediaani -annos määränpäähän oli 150 IU / kg / viikko. Tavoitteen saavuttaneista potilaista 90% teki niin 3 kertaa viikossa.
52 viikon jälkeen 57% potilaista jäi tutkimukseen, ja he saivat keskimääräisen annoksen 200 IU / kg / viikko.
05.2 Farmakokineettiset ominaisuudet
Imeytyminen
Ihonalaisen annon jälkeen epoetiini alfa -pitoisuudet huipussaan 12-18 tuntia annon jälkeen. Kerääntymistä ei tapahtunut, kun ihon alle annettiin useita 600 IU / kg viikoittaisia annoksia.
Ihon alle injektoidun epoetiini alfan absoluuttinen hyötyosuus on noin 20% terveillä koehenkilöillä.
Jakelu
Keskimääräinen jakautumistilavuus on 49,3 ml / kg 50 ja 100 IU / kg laskimonsisäisen annon jälkeen terveillä koehenkilöillä. Kroonista munuaisten vajaatoimintaa sairastavilla potilailla laskimoon annetun epoetiini alfan jakautumistilavuus vaihteli välillä 57-107 ml / kg kerta-annoksen jälkeen (12 IU / kg) ja 42-64 ml / kg toistuvan annoksen jälkeen (48-192 IU / kg). Näin ollen jakautumistilavuus on hieman suurempi kuin plasman tilavuus.
Eliminaatio
Epoetiini alfan puoliintumisaika useiden annosten laskimonsisäisen annon jälkeen on noin 4 tuntia terveillä koehenkilöillä ja puoliintumisaika ihonalaisen annon jälkeen arvioidaan noin 24 tunnilla terveillä koehenkilöillä.
Keskimääräinen CL / F -arvo 150 IU / kg 3 kertaa viikossa ja 40 000 IU kerran viikossa terveillä koehenkilöillä oli 31,2 ja 12,6 ml / h / kg. Keskimääräinen CL / F -arvo anemiaa sairastavilla 150 IU / kg 3 kertaa viikossa ja 40 000 IU kerran viikossa oli vastaavasti 45,8 ja 11,3 ml / h / kg. Suurimmalla osalla syklisen solunsalpaajahoitoa saaneista aneemisista syöpäpotilaista CL / F oli pienempi ihonalaisten 40 000 IU kerran viikossa ja 150 IU / kg 3 kertaa viikossa annettujen annosten jälkeen verrattuna terveiden henkilöiden arvoihin.
Lineaarisuus / epälineaarisuus
Terveillä koehenkilöillä havaittiin epoetiini alfan seerumipitoisuuksien suhteellista nousua annokseen 150 ja 300 IU / kg laskimonsisäisen annon jälkeen 3 kertaa viikossa. Yksittäiset ihonalaiset annokset 300-2400 IU / kg epoetiini alfaa johtivat lineaariseen suhteeseen keskimääräisen C: n ja annoksen sekä keskimääräisen AUC: n ja annoksen välillä. Käänteinen suhde havaittiin puhdistumaannos terveillä koehenkilöillä.
Tutkimuksissa, joissa tutkittiin annosten välisen ajan pidentämistä (40 000 IU kerran viikossa ja 80 000, 100 000 ja 120 000 IU joka toinen viikko), havaittiin lineaarinen mutta annoksesta riippumaton suhde keskimääräisen Cmax: n ja annoksen el "Keskimääräinen AUC ja annos välillä vakaassa tilassa" .
Farmakokineettinen / farmakodynaaminen suhde
Epoetiini alfalla on annoksesta riippuvainen vaikutus hematologisiin parametreihin, joka on riippumaton antotavasta.
Pediatriset potilaat
Kroonista munuaisten vajaatoimintaa sairastavilla lapsipotilailla on raportoitu puoliintumisajan olevan noin 6,2-8,7 tuntia epoetiini alfan moninkertaisen laskimonsisäisen annon jälkeen.
Munuaisten vajaatoiminta
Kroonista munuaisten vajaatoimintaa sairastavilla potilailla laskimoon annettavan epoetiini alfan puoliintumisaika on hieman pidempi, noin 5 tuntia, verrattuna terveisiin henkilöihin.
05.3 Prekliiniset tiedot turvallisuudesta
Toistuvan annoksen toksikologisissa tutkimuksissa koirilla ja rotilla, mutta ei apinoilla, epoetiini alfa -hoitoon liittyi luuydinfibroosin subkliininen tila. Luuytimen fibroosi on kroonisen munuaisten vajaatoiminnan tunnettu komplikaatio ihmisillä, ja se voi liittyä sekundaariseen hyperparatyreoosiin tai johtua tuntemattomista tekijöistä. Tutkimuksessa, jossa oli mukana hemodialyysipotilaita, jotka saivat epoetiini alfaa 3 vuoden ajan, luuydinfibroosin ilmaantuvuus ei ollut korkeampi kuin verrokkidialyysipotilailla, joita ei hoidettu epoetiini alfalla.
Epoetiini alfa ei aiheuta geenimutaatioita bakteereissa (Ames -testi), kromosomipoikkeavuuksia nisäkässoluissa, mikrotumisia hiirissä eikä geenimutaatioita HGPRT -lokuksessa.
Pitkäaikaisia karsinogeenisuustutkimuksia ei ole tehty. Ristiriitaiset tulokset kirjallisuudessa tietojen perusteella in vitro ihmisen kasvainnäytteistä, viittaavat siihen, että erytropoietiinit voivat vaikuttaa kasvaimen lisääntymiseen, mikä on epävarmaa kliinisessä käytännössä.
Ihmisen luuydinsolujen soluviljelmissä epoetiini alfa stimuloi spesifisesti erytropoieesia eikä aiheuta leukopoieesia Epoetiini alfan sytotoksista aktiivisuutta ei ole havaittu luuytimen soluissa.
Eläinkokeissa epoetiini alfan on osoitettu alentavan sikiön painoa, hidastavan luutumista ja lisäävän sikiön kuolleisuutta, kun sitä annetaan viikoittain noin 20 kertaa suositeltu viikkoannos ihmisellä. Näitä muutoksia pidetään toissijaisina äidin painon laskun vuoksi, ja merkitystä ihmisille ei tunneta terapeuttisilla annoksilla.
06.0 FARMASEUTTISET TIEDOT
06.1 Apuaineet
Polysorbaatti 80
Glysiini
Injektionesteisiin käytettävä vesi.
Natriummoniemäksinen fosfaattidihydraatti
Kaksiemäksinen natriumfosfaattidihydraatti
Natriumkloridia
06.2 Yhteensopimattomuus
Yhteensopivuustutkimusten puuttuessa lääkevalmistetta ei saa sekoittaa muiden tuotteiden kanssa.
06.3 Voimassaoloaika
18 kuukautta.
06.4 Säilytys
Säilytä jääkaapissa (2 ° C - 8 ° C). Tämä lämpötila -alue on varmistettava potilaalle antamisen ajankohtaan asti.
Säilytä alkuperäisessä pakkauksessa suojataksesi tuotetta valolta.
Älä jäädytä tai ravista.
Avohoitokäyttöön lääke voidaan ottaa jääkaapista ilman vaihtoa enintään 3 päivän ajaksi enintään 25 ° C: n lämpötilassa. Jos lääkettä ei ole käytetty tämän ajanjakson lopussa, se on hävitettävä.
06.5 Välipakkauksen luonne ja pakkauksen sisältö
0,3 ml (3000 IU) injektioneste, liuos, esitäytetyssä ruiskussa (tyypin I lasia), jossa on mäntä (teflonipinnoitettu kumi) ja neula kotelolla (kumi polypropeenipäällysteellä) ja PROTECS-neulansuoja (polykarbonaatti) kiinnitetty ruiskuun - paketti 1.
0,4 ml (4000 IU) esitäytetyssä ruiskussa (tyypin I lasi), jossa on mäntä (teflonipinnoitettu kumi) ja neula kotelolla (kumi polypropeenipäällysteellä) ja ruiskuun kiinnitetty PROTECS-neulansuoja (polykarbonaatti)-pakkaus 1.
0,5 ml (5000 IU) injektioneste, liuos, esitäytetyssä ruiskussa (tyypin I lasi), jossa on mäntä (teflonipinnoitettu kumi) ja neula kotelolla (kumi polypropeenipäällysteellä) ja ruiskuun kiinnitetty PROTECS-neulansuoja (polykarbonaatti) - paketti 1.
0,6 ml (6000 IU) injektioneste, liuos, esitäytetyssä ruiskussa (tyypin I lasi), jossa on mäntä (teflonipinnoitettu kumi) ja neula kotelolla (kumi polypropeenipinnoitteella) ja PROTECS-neulansuoja (polykarbonaatti) kiinnitetty ruiskuun - paketti 1.
0,8 ml (7000 IU) injektioneste, liuos, esitäytetyssä ruiskussa (tyypin I lasi), jossa on mäntä (teflonipinnoitettu kumi) ja neula kotelolla (kumi polypropeenipinnoitteella) ja PROTECS-neulansuoja (polykarbonaatti) kiinnitetty ruiskuun - paketti 1.
1,0 ml (10000 IU) injektioneste, liuos, esitäytetyssä ruiskussa (tyypin I lasia), jossa on mäntä (teflonipinnoitettu kumi) ja neula kotelolla (kumi polypropeenipäällysteellä) ja ruiskuun kiinnitetty PROTECS-neulansuoja (polykarbonaatti) - paketti 1.
06.6 Käyttö- ja käsittelyohjeet
Tuotetta ei saa käyttää ja se on hävitettävä:
• Jos tiiviste on rikki;
• Jos liuos on värillinen tai siinä on hiukkasia;
• Jos jäätyminen on tapahtunut tai sitä epäillään;
• Jos jääkaappi vioittuu.
Kertakäyttöinen tuote. Älä anna enempää kuin yksi annos ruiskua kohden, kun olet poistanut tarpeettoman määrän liuosta ruiskusta. Katso pakkausselosteen kohta 3. Miten EPREXiä (injektio -ohjeet) käytetään.
Esitäytetyt ruiskut on varustettu PROTECS-neulansuojalaitteella neulan tarttumisen estämiseksi. Pakkausseloste sisältää kaikki tiedot esitäytettyjen ruiskujen käytöstä ja käsittelystä turvalaitteen kanssa.
Käyttämätön lääke ja tästä lääkkeestä peräisin oleva jäte on hävitettävä paikallisten määräysten mukaisesti.
07.0 MYYNTILUVAN HALTIJA
Janssen -Cilag SpA, M.Buonarrotin kautta, 23-2003 Cologno Monzese (MI)
08.0 MYYNTILUVAN NUMERO
027015167 - "10000 IU / ML injektioneste, liuos esitäytetyssä ruiskussa" 1 ruisku, 3000 IU / 0,3 ml
027015179 - "10000 IU / ML injektioneste, liuos esitäytetyssä ruiskussa" 1 ruisku, 4000 IU / 0,4 ml
027015231 - "10000 IU / ML injektioneste, esitäytetty ruisku" 1 ruisku, jossa 5000 IU / 0,5 ml
027015243 - "10000 IU / ML injektioneste, liuos, esitäytetty ruisku" 1 ruisku, 6000 IU / 0,6 ml
027015268 - "10000 IU / ML injektioneste, esitäytetty ruisku" 1 ruisku, 8000 IU / 0,8 ml
027015181 - "10000 IU / ML injektioneste, esitäytetty ruisku" 1 ruisku, 10000 IU / 1ML
09.0 MYYNTILUVAN TAI UUDISTAMISPÄIVÄMÄÄRÄ
Toukokuu 1989
AIC: n uusiminen: 4. elokuuta 2008
10.0 TEKSTIN MUUTTAMISPÄIVÄMÄÄRÄ
08/2015